Images courtesy of PRISYM ID
Summary
The continual tightening of regulation around the packaging and labeling of medical devices has so far failed to stimulate a transformation of labeling infrastructure and approach among global device manufacturers. Understandable concerns about the cost and consistency of new technologies has convinced many companies to maintain traditional methods of labeling inspection – but their persistence with an old approach risks the potential breach of global regulations and continues to yield the same results. The annual volume of medical device recalls remains consistent. Likewise, the implications for cost, reputation and, at the worse extreme, patient safety are as significant as ever.
Data shows that more than half of all FDA recalls of medical devices since Q1 2014 have been caused by packaging and/or labeling error1. One of the most recent high-profile examples was the Class I recall of a Peripheral Infusion System2, where major mislabeling risked the incorrect use of the product by physicians and raised the potential for serious patient injury or death. It becomes clear that unlike many consumer and FMCG products, the cost of mislabeling a medical device can be measured in lives as well as dollars. Despite this, and despite clear regulations that require manufacturers to inspect every label and Instructions for Use (IFU) that accompanies a device, systems to support compliant labeling inspection are still not regarded as business-critical for many medical device manufacturers. Worryingly, a large percentage of the industry appears oblivious of FDA regulations around the quality requirements for label inspection3. With the stakes so high, finding a workable and cost-effective solution to the problem is becoming a major priority. But developing a clear vision to overcome it may be easier than many think.
The Business Challenge
FDA regulations stipulate that all medical device and pharmaceutical labels must be suitably checked for errors and accuracy. The scope of this inspection extends beyond assuring the pre-production validity of the data printed on the label – or multipage document such as an IFU – to the quality and legibility of the final print output.
The business challenge for medical device organizations, therefore, is to ensure that robust systems are in place to maintain compliance, mitigate risk and protect patient safety throughout the end-to-end labeling process, whilst at the same time maintaining cost-effective, efficient and profitable operations.
Regulatory Drivers
Label inspection is a regulatory requirement in both the US and European markets, irrespective of the size of an organization. Falling foul of the regulations can not only put a product out of specification but it also carries all the usual penalties of fines, product rework and potentially product recall. However, despite global guidelines being unambiguous, many within the industry appear unclear on the nature of FDA regulations.
A 2016 PRISYM ID survey of medical device and pharmaceutical organizations revealed that almost two thirds of respondents are either unsure or unaware of the FDA’s regulations on the quality system requirements for label inspections3.
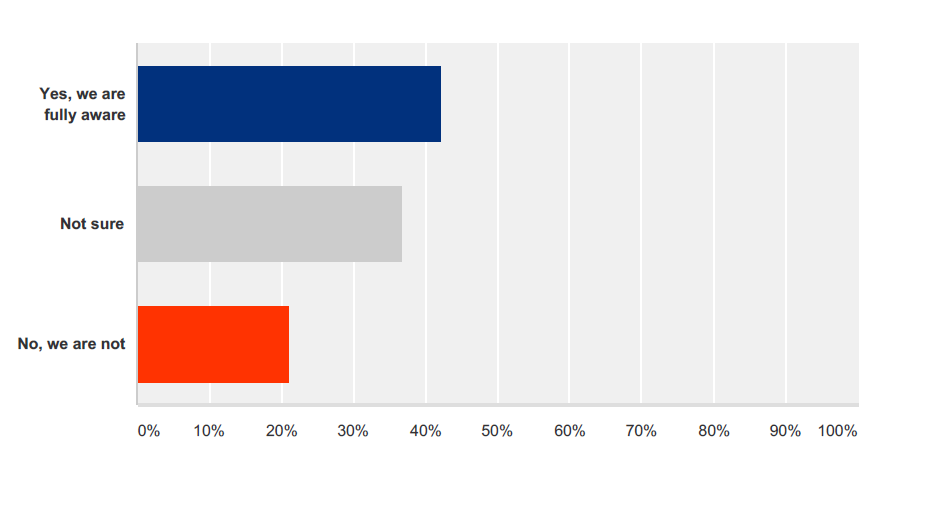
Are you aware of the FDA’s regulations on the quality system requirements for label inspections?
These requirements are outlined across a number of regulations. For example, FDA 21 CFR820.80b deals with Quality System Regulation for medical devices. It talks about the requirement for ‘incoming product’ to be inspected. In this instance, incoming product is anything that arrives from another organization and includes labeling:
FDA 21 CFR820.80b – Receiving acceptance activities. Each manufacturer shall establish and maintain procedures for acceptance of incoming product. Incoming product shall be inspected, tested, or otherwise verified as conforming to specified requirements. Acceptance or rejection shall be documented.
FDA 21 CFR820.120 is specific to device labeling. The guideline sets out a range of regulations to ensure manufacturers establish and maintain procedures to control labeling activities. These include label integrity, storage, operations and control numbers. Clause B focuses on labeling inspection:
FDA 21 CFR820.120 – Labeling inspection. Labeling shall not be released for storage or use until a designated individual(s) has examined the labeling for accuracy including, where applicable, the correct unique device identifier (UDI) or universal product code (UPC), expiration date, control number, storage instructions, handling instructions, and any additional processing instructions. The release, including the date and signature of the individual(s) performing the examination, shall be documented in the DHR.
FDA 21 CFR211.122, subpart G, deals with packaging and labeling control. It stipulates the need for written procedures to document the receipt, identification, storage, handling, sampling, examination and testing of labeling materials. It also details the requirement to maintain a robust audit trail of labeling activities, both approved and rejected. More specifically, clause G parts 2-4 highlight the importance of 100% inspection:
FDA 21 CFR211.122g – … labeling operations shall include one of the following special control procedures:
(1) Dedication of labeling and packaging lines to each different strength of each different drug product;
(2) Use of appropriate electronic or electromechanical equipment to conduct a 100-percent examination for correct labeling during or after completion of finishing operations; or
(3) Use of visual inspection to conduct a 100-percent examination for correct labeling during or after completion of finishing operations for hand-applied labeling. Such examination shall be performed by one person and independently verified by a second person.
Finally, the EU is not immune to labeling inspection. EU Directive 2001/83/EC, article 65, outlines how, in consultation with Member States, the Commission shall draw up and publish detailed guidance concerning a range of medical/healthcare products. These include:
• The wording of certain special warnings for certain categories of medicinal products;
• The particular information needs relating to non-prescription medicinal products;
• The legibility of particulars on the labeling and package leaflet;
• The methods for the identification and authentication of medicinal products;
• The list of excipients which must feature on the labeling of medicinal products and the way in which these excipients must be indicated.
In summary, the scope and detail of global regulations underline that the 100% inspection of labels in the production, manufacture and distribution of medical devices is a requirement not an option. To underline the point, the FDA has already begun to issue warning letters to organizations that have fallen foul of its quality system regulations. In 2014, 48 warning letters were issued relating to breaches of CFR820.80 and an additional four were issued regarding CFR820.1204. These notices, quite literally, serve as a warning that companies must set in place robust mechanisms and processes to manage risk and maintain compliance.
Current Methodologies
The nature of the regulations, not least FDA 21 CFR211.122g which allows provision for human and automated inspection processes, has led to a divergence in approach across the medical device sector. However, as label content becomes more complex and evolves to include variables such as local languages and country-specific requirements, companies are growing increasingly concerned that their labeling infrastructure does not sufficiently protect them against error.
PRISYM ID’s 2016 survey reveals that almost two thirds of respondents do not do enough to maintain compliance with FDA regulations (see the figure below).
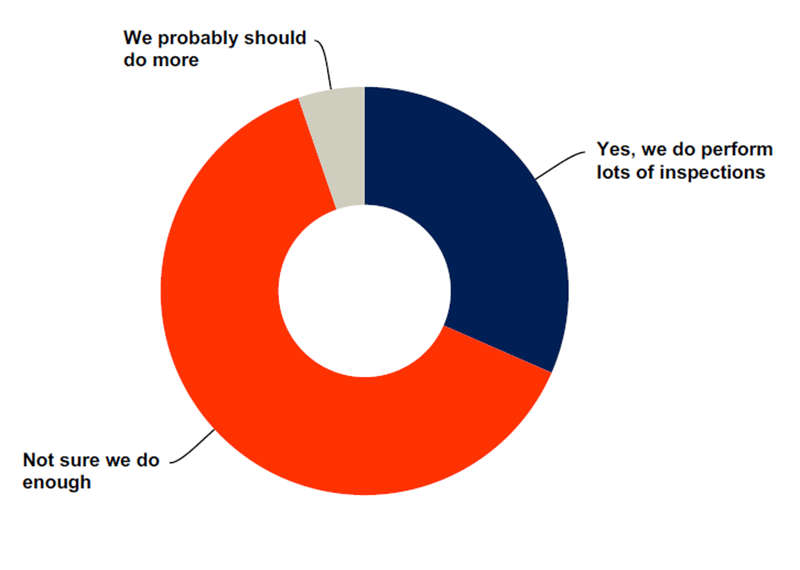
Do you feel your organization is currently conducting sufficient label inspections to be compliant with FDA regulations? (Data collected from survey conducted by PRISYM ID in 2016)
In particular, many companies believe that they could do more to monitor, identify, capture and collate errors that occur at the crucial print stage. This is an important distinction. Whilst organizations invest a large amount of time and resource in assuring pre-production data integrity, many fail to apply the same rigor to inspection once an operator has pressed print.
Companies typically focus heavily on validating data, configuring formats, managing workflow and assuring version control in the pre-print environment, but pay insufficient attention to the final print output itself. Making the assumption that a job will print in the way it was intended is ill-judged, not least because a variety of unforeseen mishaps can inadvertently impair the print process.
One of the most common is print driver conflict. This can lead to resized images, missing text, absent regulatory images or alien character sets. In addition, material quality can be hugely destructive; the topcoat of a label can deteriorate through a production run, or a print ribbon may crease, be the wrong thickness or coated incorrectly so it starts to lose print. Thirdly, mechanical issues with the print hardware can also cause unforeseen problems; a print head may be down or roller may be uneven – leading to print inconsistency or illegibility.
The potential for error during the print process is therefore significant. What’s more, it can seriously undermine earlier efforts to secure data integrity in the pre-production setting. It is perhaps not surprising that organizations believe they could do more to assure regulatory compliance in this key area.
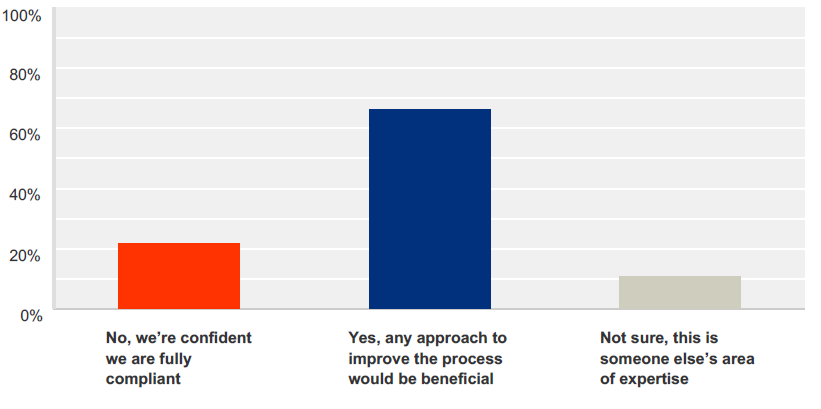
Do you feel you should do more to ensure compliance on label printing errors? (Data collected from survey conducted by PRISYM ID in 2016)
Only 21% of respondents to PRISYM’s 2016 survey claimed to be confident that their labeling operations were fully compliant3. Almost 70% said that any efforts to improve the process would only be beneficial3 (see survey results in the above figure).
At present, the ways in which companies approach labeling inspection tend to fall into two distinct camps; manual and standalone inspection.
These include (though are not limited to):
Manual Inspection
1) Pre-label and post-label inspection
One of the most common approaches is pre-and-post label inspection – 44% of respondents to PRISYM ID’s 2016 survey have adopted this methodology3. Companies to have an individual inspect the first and last labels of a run, pre-production and post-print, and maintain clear documentation to show that these passed inspection. If both inspections meet requirements, the general assumption is that everything in between is okay – and the inspection is signed-off as complete. Pre-and-post-label inspection constitutes a high-risk risk assessment.
2) Random inspection
This approach sees the company pause the print process part-way through a run and randomly inspect a label. The inspected label is once again documented in a book and available as evidence in an audit. This process focuses only on a handful of labels and does not meet the FDA requirement of 100% inspection. Despite this, it is still widely adopted. Once again, it is high-risk.
3) 100% manual inspection
This approach is labor-intensive and expensive, yet still vulnerable to human error. There are a number of variants to this approach but the most common is for an individual to sit at the printer and visually inspect every single label as it moves through the machine. Beyond the cost of labor, the flaws in this method are self-evident; it is physically impossible for a human to inspect huge volumes of detailed data, which is typically around 4-5 inches per second, and to pick out any minor fault. For example, a pin going through a thermal head can cause a barcode to be misread. It is unlikely that a human eye will be able to identify that. Moreover, the process is dependent on the individual’s attention span. Finally, should a discrepancy be detected, the individual must also be able to document it accurately. Given the speed and scale of most label runs, achieving all this appears, at best, optimistic. Yet despite these obvious risks, 100% manual inspection through the print process remains a popular approach. A third of PRISYM ID’s 2016 survey respondents adopt this methodology3.
4) 100% manual inspection, post-print
Another popular variant on 100% manual checking is for inspection to take place after a print job is complete. This approach involves moving finished labels to an inspection table or rewind unit and scrutinizing them for discrepancies and failures. This is not only time-consuming but it is also reliant on individuals to detect a discrepancy and judge it according to their company’s definition of ‘failure’. Once again, individual attention spans pose a further risk. With many print runs yielding thousands of labels, these are perhaps unrealistic expectations to believe a human can deliver infallible results.
5) Manual template
This approach is based on companies creating a label template prior to production and then laying it on top of finished labels to establish whether or not there is content or gaps in the appropriate spaces. It’s a primitive method but it’s surprisingly common amongst smaller medical device organizations.
Standalone Vision inspection*
An alternative method is to adopt a ‘standalone’ approach to label inspection, whereby the print inspection system is entirely separate from the label management system. The approach relies on using a standalone application – a ‘vision system’ – to design a label template in line with individual company specifications, then using the template to form an inspection mask that supports automated checking post-print. This is a huge step forward for manual processes. Good standalone applications can be very effective, helping businesses marry customized label standards with print outputs to enable more accurate inspection. Moreover, the approach allows companies to check every single label.
However, whilst vision systems have proved very effective in commercial pharmaceutical manufacturing where there are large production runs and limited data to inspect, their use can be more challenging for medical device companies. In the device sector, print runs are often smaller but labeling data and designs are much more complex. As such, the expertise and time required to configure a vision inspection application to identify all errors has often proved prohibitive, resulting in many technology trials failing an operational ROI assessment.
As companies use label management software and vision inspection systems that are supplied as separate systems, from separate suppliers, this standalone approach treats inspection and label management as two silo activities. The lack of integration is problematic. It is not only inefficient and increases risk throughout the label lifecycle, but it also leaves companies with two individual audit logs; one for printing, the other for inspection.
Whilst all of the above methods can be used as a risk assessment to determine whether finished labels are accurate, they each have limitations – some more substantial than others. Moreover, the vast majority fail to meet the basic requirement of 100% inspection and leave companies exposed to risk. As the FDA turns up the heat on medical device organizations, large and small, to demonstrate compliance, the industry needs a more effective way to mitigate risk. Companies need to find a solution that gives them end-to-end label tracking and the ability to understand that what was sent to the printer not only physically printed, but can also be proven in a single audit log. For more effective joined-up thinking, an integrated approach is the only solution. However, until now, an integrated solution has not been available.
A Solution
One example of a solution to these challenges is the fully integrated label software and print inspection system developed by PRISYM ID.
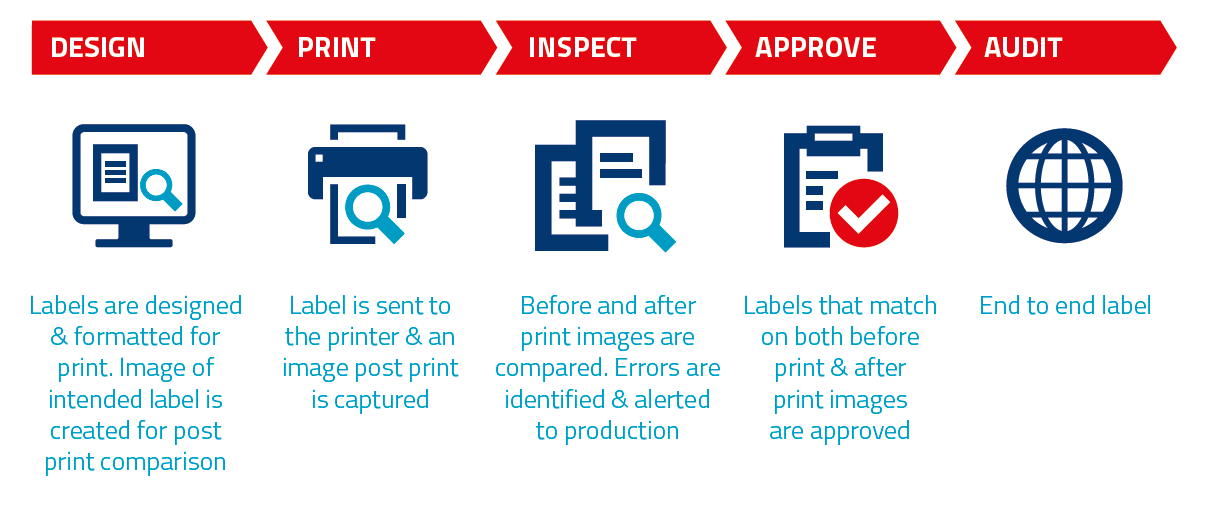
PRISYM ID’s fully integrated label software and print inspection solution.
PRISYM 360 Vision, a module for PRISYM 360 label management software, provides complete print inspection set-up, test and management functionality. Its Vision Inspection Wizard automatically creates inspection masks from label designs, reducing the effort and expertise required for configuration and testing. Moreover, since it is fully integrated with the industry-leading in-line print verification system, Crest Solutions’ PrintInspectorTM, it successfully marries label management and print inspection, facilitating the automated inspection of every label using the design and content sent to print.
Crucially, as the labeling infrastructure of medical device organizations becomes increasingly scrutinized, the solution is underpinned by an integrated audit log to document a single source of the truth. PRISYM 360 Vision not only helps companies assure FDA compliance, it minimizes operational costs, increases throughput and efficiency and significantly mitigates the risk of error throughout the labeling process.
Conclusion
The medical device industry spends huge amounts of time ensuring its datasets are accurate, integrated, designed and approved for compliant labeling outputs. However, few companies spend sufficient time ensuring that the label that emerges at the end of the process is printed correctly. The repercussions of this could potentially be severe. As the regulatory climate becomes ever more challenging, global organizations must do all that they can to ensure that they have optimal labeling systems and processes to meet the demands of the FDA and European regulators. This requires cross-functional, organization-wide collaboration to assure that data formats, integration points and production infrastructure are all aligned – and are underpinned by a system that delivers that single source of the truth.
In the highly regulated environment of medical devices, label lifecycle management is without doubt a business-critical system. It should be treated as such. As the regulators circle above, perhaps it’s time your business developed a clear vision. PRISYM 360 Vision: a groundbreaking leap into a new world for label lifecycle management.
References
1. Stericycle ExpertSOLUTIONS Quarterly Index, www.stericycleexpertsolutions.com/top-5-causes-of-medical-device-recalls/
2. FDA, Medical Device Recalls www.fda.gov/MedicalDevices/Safety/ListofRecalls/ucm433765.htm
3. Prisym ID, Labeling Quality, Industry Survey 2016
4. FDA, 2014 Annual FDA Medical Device Quality System Data
* Separate label management and print inspections systems



