The FENIX Continence Restoration System
The U.S. Food and Drug Administration today (18 December) approved the Fenix Continence Restoration System to treat fecal incontinence in patients who are not candidates for, or have previously failed, medical or other surgical options.
Fecal incontinence is the inability to control bowel movements. It is a common problem that is frequently underreported, especially among older adults. The most common cause of fecal incontinence is damage to the muscles around the anus (anal sphincter) from vaginal childbirth or functional disorders such as diabetes.
“Non-invasive treatment options for fecal incontinence, such as drugs, dietary changes and other medical measures, sometimes don’t adequately address a patient’s symptoms,” said William Maisel, M.D., M.P.H., acting director of the Office of Device Evaluation in the FDA’s Center for Devices and Radiological Health. “The Fenix System affords a viable surgical option to address this condition when other methods have failed to improve a patient’s quality of life.”
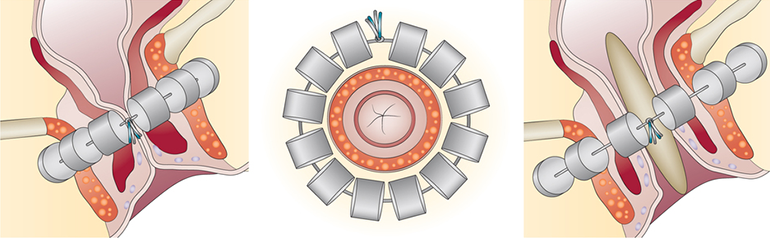
The Fenix System has three components: an implant, an anal sphincter sizing tool and an introducer tool. The implant is a series of titanium beads with magnetic cores that are connected by titanium wires to form a ring shape. The attractive force of the magnetic beads augments the anal sphincter to minimize involuntary opening of the anal canal, reducing the likelihood of severe fecal incontinence. The implant device is offered in multiple sizes to accommodate variation in sphincter size. The sizing tool is used to associate the anal sphincter size to an appropriate implant. The introducer tool is used to guide the sizing tool and the implant into position.
The FDA reviewed data for the Fenix System through the humanitarian device exemption (HDE) process. A Humanitarian Use Device (HUD) is a device that is intended to benefit patients by treating or diagnosing a disease or condition that affects or is manifested in fewer than 4,000 individuals in the U.S. per year. Since a device manufacturer`s research and development costs could exceed its market returns for diseases or conditions affecting small patient populations, the HUD provision of FDA regulations provides an incentive for the development of devices for use in the treatment or diagnosis of diseases affecting these populations.
The Fenix System was studied in 35 adults who failed conventional medical therapy for treating fecal incontinence. Study evaluations were performed before and after the procedure and at six weeks, three months, six months and 12 months post-implant. The 15 participants studied at U.S. sites will have annual evaluations until five years after the device was implanted.
Probable benefits were evaluated using a bowel diary to track fecal incontinence events and by a validated, disease-specific questionnaire called the Fecal Incontinence Quality of Life Scales to quantify changes in quality of life before and after implantation of the Fenix System. After 12 months, 62.9 percent of participants experienced a reduction of fecal incontinence episodes by half or more; 54.3 percent experienced a reduction in fecal incontinence days by half or more; and 37.1 percent experienced a reduction in urgent episodes by half or more. Study participants also showed improvements in quality of life measures including improvements in depression, self-perception, and feelings of embarrassment. These results suggest that patients with fecal incontinence could benefit from the device when they have failed other fecal incontinence therapies.
The Fenix System should not be implanted in patients with suspected or known allergies to titanium. The sizing tool and introducer tool should not be used in patients with suspected or known allergies to titanium, stainless steel, nickel or ferrous materials.
The implant is considered magnetic resonance unsafe. After implantation, patients should not be exposed to a magnetic resonance imaging (MRI) environment as it could interfere with the magnetic strength and the function of the device.
A recommendation should be made to patients receiving the Fenix System to register their implant with the MedicAlert Foundation or an equivalent organization. In the event alternative diagnostic procedures cannot be used and MRI is required, the implant can be safely removed.
Adverse events identified in the clinical trial for the Fenix System include pain, infection, impaction or defecatory disorder, device erosion, device removal/re-operation, and bleeding.
The Fenix System is manufactured by Torax Medical Inc., based in Shoreview, Minnesota.