As manufacturers are preparing to include unique device identifiers (UDIs) on more of their products this fall, the Food and Drug Administration (FDA) has provided additional guidance on the content and form of these identification tags—and is seeking feedback on its proposal.
The UDI system was developed in an effort to better track medical devices throughout their life cycle. The FDA’s UDI rule, finalized in September 2013, will eventually require every medical device label and packaging to include a UDI, with some exceptions. The FDA is phasing in these requirements over seven years, with requirements for Class II medical devices—which are those associated with moderate risk—set to be implemented in September. According to the agency, the UDI must include a “device identifier” that indicates the version or model of the device and at least one “production identifier,” such as the batch number, the expiration date, or the date the device was manufactured.
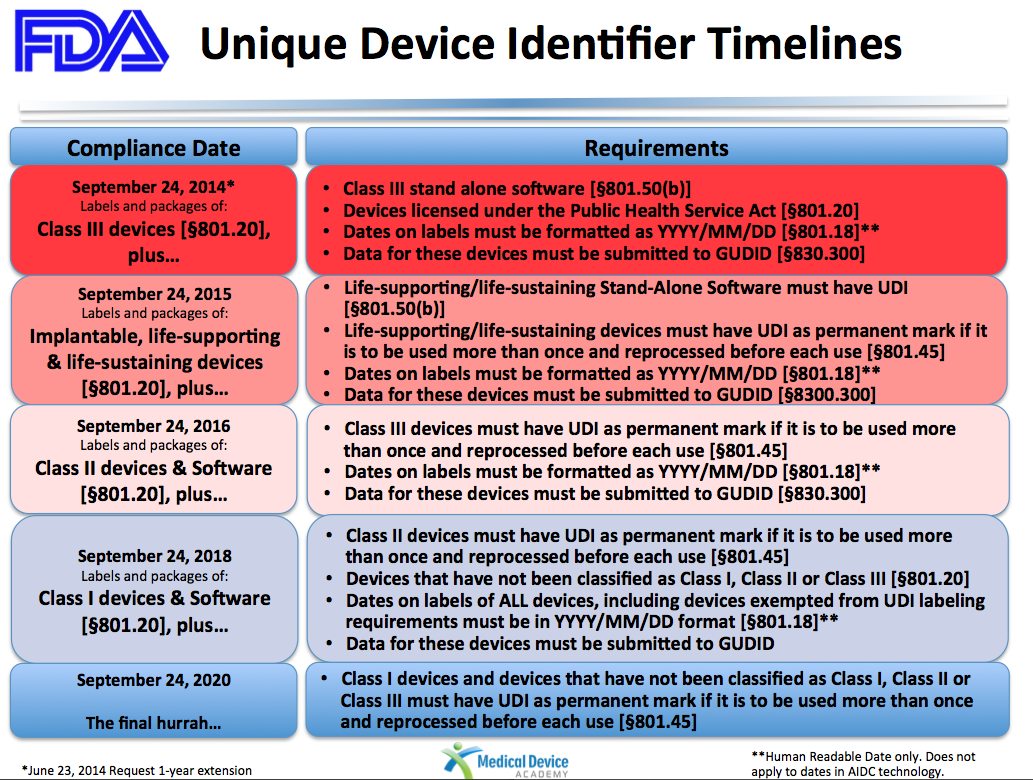
Image: FDA
However, meeting the FDA’s UDI requirements is not as simple as slapping the required information onto a label. The new draft guidance stipulates that the UDI must be presented in two formats: an “easily readable plain-text form” and a machine-readable version that can be scanned by a barcode reader or another device so it can be automatically added to a patient’s electronic health record or other computer system.
The guidance also specifies how this information should be ordered. For example, the device identifier should come before the production identifiers, and the plain text UDI should be positioned below or near the machine-readable version. The FDA also states that UDI information should precede non-UDI information.
If space is limited, the FDA will allow both versions of the UDI to be split over multiple lines.
Comments and suggestions on the draft guidance can be submitted online at www.regulations.gov until Sept. 26.
– See more at: http://www.aami.org/newsviews/newsdetail.aspx?ItemNumber=3757#sthash.rVALxuRT.dpuf



