It sounds like something out of the Borg in Star Trek. Nano-sized robots self-assemble to form biological machines that do the work that keeps one alive. And yet something like this really does go on.
Every cell in our body – be they flesh and blood, brain and everything in between – has identical DNA, the twisted staircase of nucleic acids uniquely coded to each organism. Complex assemblages that resemble molecular machines take pieces of DNA called genes and make a brain cell when needed, instead of, say, a bone cell. These molecular machines are so complex, yet so tiny, that scientists today are just starting to understand their structure and function using the latest microscopes and supercomputers. Biological molecular machines could lay the foundation for developing cures to diseases like cancer. How small can one see, and what will one find?
Cryo-electron microscopy combined with supercomputer simulations have created the best model yet, with near atomic-level detail, of a vital molecular machine, the human pre-initiation complex (PIC). A science team from Northwestern University, Berkeley National Laboratory, Georgia State University, and UC Berkeley published their results on the PIC May 2016 in the journal Nature.
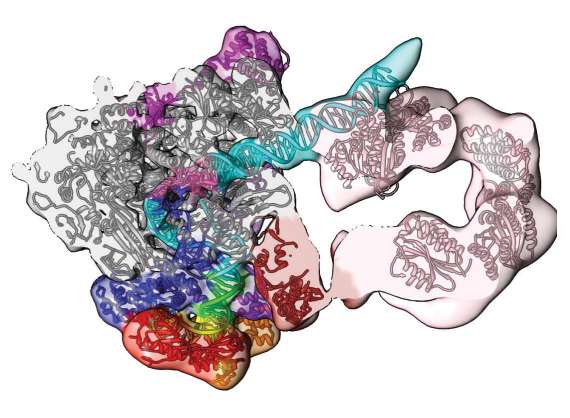
Supercomputing and cryo-electron microscopy reveal this section of the human pre-initiation complex. The open conformation density map and model show the path of the DNA (blue/green) and its engagement by the transcription factor component TFIIH (pink). Reprinted by permission from Macmillan Publishers Ltd: He, Y. et al. Near-atomic resolution visualization of human transcription promoter opening. Nature 533, 359–365 (2016). (Credit: Phys.org)
“For the first time, structures have been detailed of the complex groups of molecules that open up human DNA,” said study co-author Ivaylo Ivanov, associate professor of chemistry at Georgia State University. Ivanov led the computational work that modeled the atoms of the different proteins that act like cogs of the PIC molecular machine.
The PIC finds genes associated with making a specific protein, such as an antibody or an enzyme. There the PIC pulls apart the two strands of DNA and feeds the coding strand to the workhorse enzyme RNA polymerase II. This starts transcription, where DNA bits are copied by RNA polymerase II into a single strand of messenger RNA. The RNA makes its way to ‘protein factories’ in the cell called ribosomes that take them as orders for which protein to make. If DNA is like the blueprint of a new house, RNAs are instructions to the ‘contractors’ at the ribosome work station. The manufactured proteins are like the nails, wood, plaster, and just about everything else in the house.
The experiment began with images painstakingly taken of PIC. They were made by a group led by study co-author Eva Nogales, a professor in the Department of Molecular and Cellular Biology at UC Berkeley and also Senior Faculty Scientist at the Lawrence Berkeley National Laboratory and Howard Hughes Medical Investigator.
Nogales’ group used cryo-electron microscopy (cryo-EM), a rising star in lab techniques. They cryogenically froze human PIC bound to DNA. The freezing kept it in a chemically-active, near-natural environment. Next they zapped it with electron beams. Thanks to recent advances in direct electron detector technology, cryo-EM can now image at near atomic resolution large and complicated biological structures that have proven too difficult to crystalize. The go-to technique, X-ray crystallography, requires crystallized specimens, and cryo-EM avoids this hard step.
Over 1.4 million cryo-EM ‘freeze frames’ of PIC were processed using supercomputers at the National Energy Research for Scientific Computing Center to sift out background noise and reconstructed three-dimensional density maps that show details in the shape of the molecule that had never been seen before.
“Cryo-EM is going through a great expansion as are all the computer software used to generate both the density maps and also to interpret them like we’ve done in this study,” Nogales said. “It is allowing us to get higher resolution of more structures in different states so that we can describe not just one picture of how they look, but several pictures showing how they are moving. We don’t see a continuum, but we see snapshots through the process of action.”
Study scientists next built an accurate model that made physical sense of the density maps of PIC using XSEDE, the eXtream Science and Engineering Discovery Environment, funded by the National Science Foundation. XSEDE allows scientists to interactively share computing resources, data and expertise via a single virtual system. Ivaylo Ivanov’s team has run over four million core hours of simulations on the Stampede supercomputer at the Texas Advanced Computing Center to model complex molecular machines, including those for this study. Ivanov’s broader molecular machine work also includes an XSEDE allocation of 1.7 million core hours on the Comet supercomputer at the San Diego Supercomputing Center.
“I have been using XSEDE resources for more than 12 years now,” Ivanov said. “Without the availability of XSEDE resources, all of our research would have been much more limited in terms of the systems that we can address. For us, XSEDE has been absolutely essential.”
The goal of all this computational effort is to produce atomic models that tell the full story of the structure and function of the protein complex of molecules. To get there Ivanov’s team took the twelve components of the PIC assembly and created homology models for each component that accounted for their amino acid sequences and their relation to similar known protein 3-D structures.
Next they approximated the experimental densities Nogales’ team found onto a grid. “We can use a method called molecular dynamics flexible fitting,” explained Ivanov, “where you essentially run a molecular dynamics simulation. And you use the experimental density to bias the atoms in the molecular dynamics simulation to move into the denser regions of the EM map. That’s the process of flexible fitting to the EM map.”
They refined the model with the Phoenix crystallographic refinement package. “That is a complimentary technique that allows us to position side chains and improve the model so that we can capture all the details that are present in the density map,” Ivanov said.
XSEDE was “absolutely necessary” for this modeling, said Ivanov. “When we include water and counter ions in addition to the PIC complex in a molecular dynamics simulation box, we get the simulation system size of over a million atoms. One cannot run that on a work station or even on a modest cluster. For that we really need to go to a thousand cores. In this case, we went up to two thousand and forty-eight cores. And for that we needed access to Stampede,” Ivanov said.
One of the insights gained in the study is a working model of how PIC opens the otherwise stable DNA double helix for transcription. Nogales explained that one could imagine a cord made of two threads twisted around each other. Hold one end very tightly. Grab the other and twist it in the opposite direction of the threading to unravel the cord. That’s basically how the living machines that keep us alive do it.
“The DNA needs to be opened and moved into the polymerase active site to encode for the first RNA nucleotide,” explained Nogales. “The pre-initiation complex is holding the two strands of the DNA very tightly together at one end, so that they cannot move and they cannot open. On the other side of the PIC there is a machine that uses energy to push the DNA, twisting it in the opposite direction in which the two strands are threaded. And when this happens, in between the two sides, the strands will open,” said Nogales.
This study resolved the structure of that molecular machine that acts like the twisting fingers, the transcription factor component TFIIH. “TFIIH has a translocase sub-unit, whose role is to simultaneously push the DNA toward the active site of the polymerase and unwind the DNA. By the combined pushing and unwinding, effectively you are separating the two strands of the DNA,” Ivanov said.
Both scientists said that they are just beginning to get an atomic-level understanding of transcription, crucial to gene expression and ultimately disease. “Many disease states come about because there are errors in how much a certain gene is being read and how much a certain protein with a certain activity in the cell is present,” Nogales said. “Those disease states could be due to excess production of the protein, or conversely not enough. It is very important to understand the molecular process that regulates this production so that we can understand the disease state.”
“This work illustrates well two general principles that will drive science in the next few years,” commented Peter Preusch, program officer with the National Institutes of Health (NIH). “One is the application of hybrid methods – combinations of biophysical methods including x-ray crystallography and cryoEM along with large scale computational methods to integrate information on larger molecular complexes. Two, there’s the requirement for team science drawing the expertise of multiple investigators to solve problems that cannot be tackled by any single lab working alone.” Peter Preusch is the Biophysics Branch Chief, Cell Biology and Biophysics Division, National Institute of General Medical Sciences, NIH.
While this fundamental work does not directly produce cures, it does lay the foundation to help develop them in the future, said Ivanov. “In order to understand disease, we have to understand how these complexes function in the first place… A collaboration between computational modelers and experimental structural biologists could be very fruitful in the future. “
The May 2016 Nature Articles study (DOI: 10.1038/nature17970), “Near-atomic resolution visualization of human transcription promoter opening,” was authored by Yuan He, Lawrence Berkeley National Laboratory and now at Northwestern University; Chunli Yan and Ivaylo Ivanov, Georgia State University; Jie Fang, Carla Inouye, Robert Tjian, Eva Nogales, UC Berkeley. Funding came from the National Institute of General Medical Sciences (NIH) and the National Science Foundation.



