Biological small-angle X-ray scattering (SAXS) is an experimental technique that provides low-resolution structural information on macromolecules. The surge of popularity of the technique is a result of recent improvements in both software and hardware, allowing for high-throughput data collection and analysis, reflected in the increasing number of dedicated SAXS beamlines such as BM29 at the ESRF, P12 at EMBL Hamburg and B21 at Diamond Light Source.
However, as for most other macromolecular structural techniques, radiation damage is still a major factor hindering the success of experiments. The high solvent proportion of biological SAXS samples means that hydroxyl, hydroperoxyl radicals and hydrated electrons are produced in abundance by the radiolysis of water when it is irradiated with X-rays. These radicals can then interact with the protein molecules, ultimately leading to protein aggregation, fragmentation or unfolding. Furthermore, molecular repulsion due to protein charging can also decrease the scattering at low angles.
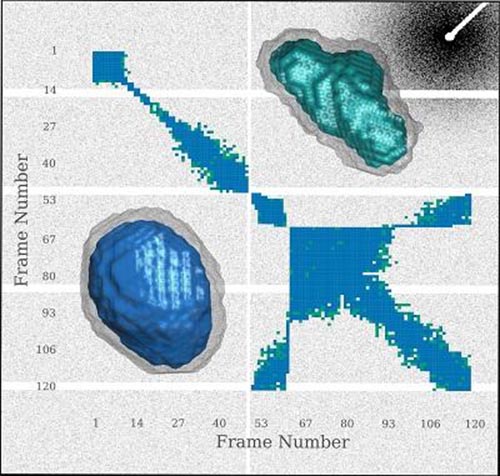
SAXS envelopes of Glucose Isomerase generated from frames collected early (bottom left) and late (top right) in the experiment. (Credit: Brooks-Bartlett et al)
Common methods used to reduce radiation damage to biological SAXS samples are generally concerned with limiting the X-ray exposure to any given volume of sample. In an analogous manner, cryo-cooling samples down to 100 K for SAXS (cryoSAXS) has been reported to increase the dose tolerance of SAXS samples by at least two orders of magnitude.
Applications of the above radiation damage mitigation approaches are unable to completely circumvent its detrimental effects, in particular the change of the scattering profile throughout the experiment. It is necessary to determine whether any two scattering profiles are similar so that noise can be reduced by averaging over similar curves.
For experiments by different researchers to be inter-comparable, the progression of radiation damage is most usefully tracked as a function of the dose absorbed by the sample. RADDOSE-3D is a free and open source software program used to calculate the time- and space-resolved three-dimensional distribution of the dose absorbed by a protein crystal in a macromolecular crystallography experiment; however, there is no equivalent software available for SAXS. Radiation damage studies in SAXS thus currently require the experimenters to correctly parameterize the experiment and manually calculate a single estimate of the dose within the sample.



