
In April 2014, Fred Upton, energy and commerce committee chairman, partnered with Rep. Diana DeGette to conduct a comprehensive look at the cycle of cures, from discovery to development to delivery and back to discovery.
It has been reported that among the 10,000 known diseases, 7,000 of which are considered rare, there are treatments for only 500. According to Dr. Francis Collins, Director of the National Institutes of Health (NIH), it now takes “around 14 years and $2 billion or more” to develop a new drug and “more than 95% of [such] drugs fail during development.”
In April 2014, Fred Upton, energy and commerce committee chairman, partnered with Rep. Diana DeGette to conduct a comprehensive look at the cycle of cures, from discovery to development to delivery and back to discovery. Over the course of the last year, the committee has had the privilege of participating in this wide-ranging conversation with patients, providers, innovators, regulators and researchers from around the country.
Countless ideas were submitted in response to the committee white papers, raised during the eight hearings convened by Joe Pitts, health subcommittee chairman, and discussed at over a dozen roundtables hosted both at the committee and by members in their districts all across the country.
After this year of listening, it is clear that Congress must take bold action to accelerate the discovery, development and delivery of promising new treatments and cures for patients and to maintain our nation’s standing as the biomedical innovation capital of the world. It is clear that America needs a strategy for 21st century cures.
The committee is circulating this discussion document, including a number of ideas proposed by both Republicans and Democrats, with the intent of continuing this national dialogue. Continued feedback is essential to this initiative’s success.
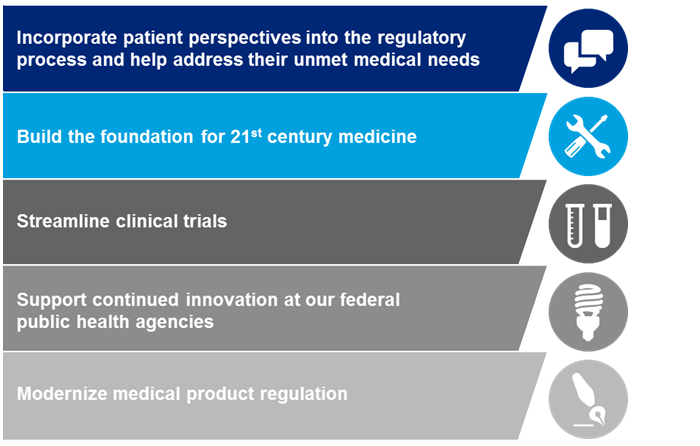
Upton looks forward to continuing to work with both Republicans and Democrats on ideas to provide additional resources, including to the National Institutes of Health. While the legislative language released is neither perfect nor complete, with the aforementioned goals in mind the committee has included provisions that would: (1) incorporate patient perspectives into the regulatory process and help address their unmet medical needs; (2) build the foundation for 21st century medicine; (3) streamline clinical trials; (4) support continued innovation at our federal public health agencies; and (5) modernize medical product regulation. The discussion is ongoing, and continued feedback in this collaborative effort is critical.

Because no one understands a particular condition or disease better than patients living with it, FDA would be required to establish a structured framework for the meaningful incorporation of patient experience data into the regulatory decision-making process, including the assessment of desired benefits and tolerable risks associated with new treatments.
Title I – Putting patients first by incorporating their perspectives into the regulatory process and addressing unmet medical needs
Provisions drafted by Pitts and Rep. Cathy McMorris Rodgers that are included in Title I would build on the Patient Focused Drug Development program at the Food and Drug Administration (FDA).
Because no one understands a particular condition or disease better than patients living with it, FDA would be required to establish a structured framework for the meaningful incorporation of patient experience data into the regulatory decision-making process, including the assessment of desired benefits and tolerable risks associated with new treatments.
Title I also includes a number of proposals that would help address patients’ unmet medical needs. In a perfect world, research and development dollars would flow toward the areas of highest need where the severity or burden of disease was most pronounced. However, as noted by the President’s Council of Advisors on Science and Technology, the current system in many ways discourages investment in therapies for scientifically complex diseases with longer development times. Collins has encouraged Congress to tackle this problem since today’s patent framework often makes it economically unviable to bring such therapies to market, even when they have shown early promise.
Title I contains a series of legislative proposals intended to address this imbalance, including the Dormant Therapies Act. This bill was introduced by Senators Orrin Hatch and Michael Bennet in December 2014 and is based on the MODDERN Cures Act, spearheaded in the House by committee member Rep. Leonard Lance and supported by 47 Democratic and 48 Republican cosponsors.
In addition, Title I includes provisions that would enhance the ability of the National Center for Advancing Translational Sciences (NCATS) at NIH to use of Other Transaction Authority and award grants and contracts for research into whether previously approved therapies could be repurposed to treat different conditions or diseases once the products’ patent life and exclusivity periods have expired.
Also included is the OPEN ACT, introduced by committee members Reps. Gus Bilirakis and G.K. Butterfield, which would reward private investment in such repurposing efforts for rare diseases, as well as other provisions intended to incentivize research on previously approved products.
Encouraging the development and approval of new antibiotics is a longstanding bipartisan priority of the committee. In the Food and Drug Administration Safety and Innovation Act (FDASIA), the committee included the GAIN Act to help stimulate investment in new infectious disease products. Title I builds on the GAIN Act with the inclusion of the Antibiotic Development to Advance Patient Treatment (ADAPT) Act, championed by committee members Reps. John Shimkus and Gene Green, and supported by an extensive bipartisan group of members. The ADAPT Act would further this progress by expediting the development and approval of these desperately needed therapies.
Regardless of the incentives in place, new FDA-approved treatments will not reach patients soon enough unless the time it takes to conduct clinical trials is significantly shortened. In addition to reducing a number of the administrative burdens involved in setting up and then running a trial, provisions included in Title I would enable sponsors to better leverage recent advancements in science and technology to weed out unsafe or ineffective compounds sooner. This will focus resources on accelerating the development and approval of treatments with the greatest potential to benefit to patients, particularly when they would address unmet medical needs.
FDA already has broad authority to review and ultimately approve new products on an expedited basis using biomarkers and surrogate endpoints that can be assessed in modern trial designs and relied on to evaluate safety and clinical benefit earlier in the process. However, the agency can only take full advantage of these authorities if it is comfortable with the evidence presented and the methodologies used to compile it.
Developed based on conversations led by committee member Rodgers, language included in Title I would establish a transparent process at FDA with specified timeframes for the development of evidentiary standards and the review and qualification of surrogate endpoints for broader utilization in regulatory decision-making. So FDA is not overburdened with requests and it can continue to focus on actual product reviews, the agency will be authorized to partner with an outside entity or entities to establish a similar process for the qualification of biomarkers not used as surrogate endpoints. In doing so, this legislation would explicitly maintain a product sponsor’s ability to confidentially discuss the use of such tools in product-specific, proprietary contexts.
In addition to containing a series of provisions that would help expedite the development and approval of safe and effective drugs for unmet needs, Title I includes a number of like-minded policies for medical devices. For example, it would authorize FDA to implement similar, though not identical, regulatory concepts that have proven successful on the drug side, while expanding the scope of valid scientific evidence FDA should consider in the review of such products.
Pursuant to a statutory framework developed by Pitts, FDA would establish a program to provide priority review for breakthrough technologies that meet certain qualifying criteria. A specifically assigned review staff, led by senior agency personnel, will collaboratively work with the sponsors of such devices to expedite their development, including early agreement on trial designs and endpoints. Should a sponsor of a device that met the criteria for priority review opt to apply for accelerated approval, FDA could approve the device upon a determination that it has an effect on a surrogate or intermediate clinical endpoint that is reasonably likely to predict clinical benefit.
For both devices and drugs, learning about the benefits and risks of a product does not end when FDA initially approves it for a population or subpopulation of patients with a certain condition or disease. Different uses for the treatment are often discovered after FDA approval, many times for different conditions and diseases. As innovative companies know more about their products than anyone, precluding them from responsibly communicating about new scientific and medical developments does not promote the public health.
FDA’s current rules and policies governing what drug and device developers may say about their own products were designed decades ago. Since then, the way that medicine is practiced and delivered and the way that information is communicated have fundamentally changed. Patients and providers are increasingly turning to the Internet for information on potential treatment options. FDA must reconsider its regulation of manufacturer communications in this space, and the agency will do so under provisions drafted by committee member Rep. Billy Long.
Further, the committee is currently working on a proposal that would clarify and rationalize these broader rules of the road so scientific and medical developments can be proactively shared with physicians, insurers and researchers, with appropriate safeguards, in order to optimize patient care.
As new uses are being discovered in the delivery phase, everyone benefits from FDA’s review of such indications as soon as possible. With that in mind, Title I includes a proposal drafted by Rep. Michael C. Burgess, M.D. that would streamline the supplemental approval process, initially for oncology indications, by enabling sponsors to submit qualified summaries of clinical data.

In addition to the work being done in industry, academia and government, there are a number of important organizations and partnerships that have supported the biomedical research enterprise across the country.
Title II – Building the foundation for 21st century medicine, including helping young scientists
The committee’s conversations over the past year have underscored that the discovery, development and delivery process is a cycle, data captured and analyzed on the delivery side informs new discoveries and better, more targeted solutions for patients. While improvements to individual components of this cycle can make a meaningful difference, the U.S. must ensure that, in its entirety, its cycle is a well-oiled, constantly revolving generator of innovative new treatments and cures.
In addition to the work being done in industry, academia and government, there are a number of important organizations and partnerships that have supported the biomedical research enterprise across the country. What is missing is a public-private partnership that brings together these various players to develop a strategic research agenda, facilitate collaboration, identify gaps and opportunities across this cycle. The partnership should also award competitive grants for activities that accelerate the discovery, development and delivery of innovative cures, treatments and preventative measures for patients in the U.S.
This legislation, led by Rodgers, would establish such a public-private partnership to be known as the “21st Century Cures Consortium” that, while broader in scope, would be modeled after the Innovative Medicines Initiative (IMI) in Europe.
Further, a Medical Product Innovation Advisory IMI is a partnership between the European biopharmaceutical industry and the European Union “working to improve health by speeding up the development of, and patient access to, innovative medicines, particularly in areas where there is an unmet medical or social need.”

Commission will be established as an agency of Congress, similar to the Medicare Payment Advisory Commission (MedPAC), to review federal policies and interactions between relevant agencies that impact various components of the cycle and annually report to Congress on the state of medical product innovation.
Establishing both the 21st Century Cures Consortium and Medical Product Innovation Advisory Commission would ensure ongoing consideration of, and improvements to, the discovery-development-delivery cycle.
The foundation for 21st century medicine cannot be built on top of decades-old structures. Genomics, data analytics, health information and other constantly evolving data sources, technologies and platforms have already fundamentally transformed many aspects of patient care and healthcare delivery. The accuracy and reliability of a number of these products and services can be a matter of life and death, which underscores the need for appropriate regulation to ensure safety and efficacy. However, using standards set in a medical device law that was enacted in 1976 may not always be a suitable solution.
Based on the Sensible Oversight for Technology Which Advances Regulatory Efficiency (SOFTWARE) Act, introduced by Reps. Marsha Blackburn, Green, Greg Walden, G.K. Butterfield and DeGette, Title II includes provisions that would authorize the development of new standards for certain types of medical software.
Following FDA’s proposed guidance altering the regulatory landscape for the review and oversight of laboratory developed tests, a broader conversation about the need to modernize the regulation of diagnostics has reached a fever pitch. The committee is encouraged by stakeholder efforts to build consensus around what a modern framework should look like and is working toward the inclusion of such a proposal.
Innovative products that harness advances made in genomics and analytics have enabled researchers and providers to discover new ways to proactively diagnose and treat patients in a more personalized manner based on their unique set of circumstances. In order to optimize patient care and collaboratively discover the next generation of patient-centered solutions, data about how different therapies are impacting patients in the real world must be shared and put to work.
Based on the leadership of Rep. Morgan Griffith, Title II includes provisions intended to establish a 21st century data sharing framework. Further, it would go a long way toward unlocking the research potential of data sitting in thousands of siloed healthcare facilities across the country and enable patients who want to play a more proactive role in finding better treatments or a cure for their disease to do so in a responsible manner that continues to protect their privacy. In order for any of this to happen at scale, data must be able to flow through the health system. Burgess is continuing efforts to build a national interoperable health information infrastructure. The committee would enthusiastically support and include a meaningful, workable proposal that would realize this vision as soon as possible.

The committee’s conversations over the past year have underscored that the discovery, development and delivery process is a cycle, data captured and analyzed on the delivery side informs new discoveries and better, more targeted solutions for patients. While improvements to individual components of this cycle can make a meaningful difference, the U.S. must ensure that, in its entirety, its cycle is a well-oiled, constantly revolving generator of innovative new treatments and cures.
Building the foundation for 21st century medicine also requires empowering our nation’s emerging scientists and researchers. The committee has heard from this younger generation about the daunting challenges they face in winning grants and pursuing their careers in research. Included in Title II are provisions from Rep. Andy Harris’ YES to Cures Act that would help address this problem and keep the most talented and promising researchers here in the U.S.

Title III – Modernizing clinical trials
Throughout the 21st Century Cures initiative, research sponsors, medical institutions and others have lamented the increasing time and costs involved in conducting clinical trials in the U.S. Like many young scientists, these trials are moving overseas to avoid the bureaucratic hurdles that are currently in place here in the U.S. Many of the rules and regulations established for the review of clinical trial protocols and the protection of participating human subjects were drafted when trials rarely involved multiple investigators across a number of different trial sites. Oftentimes, due in part to this antiquated framework, each individual entity must have its own institutional review board (IRB) approve the trial before patient recruitment can even commence. As a result, the development and assessment of potential new treatments and cures is delayed and patients are forced to wait.
Rodgers and DeGette have put forth a common-sense piece of legislation included in Title III that would significantly reduce regulatory overlap and administrative inefficiency. The Clinical Research Modernization Act would stipulate that clinical trials subject to FDA regulations should not also be subject to the duplicative—and sometimes conflicting—provisions known as the Common Rule under the Public Health Service Act. Further, it would encourage efficient IRB review by clarifying the role of “central” and local IRBs and addressing concerns about legal liability.
While cutting through this red tape would significantly help, much work remains to be done if less-expensive, more efficient trials are no longer the exception to the rule. Aided by innovative technologies and statistical modeling, broader application of adaptive trial designs would be an encouraging step in the right direction. New committee member Rep. Chris Collins has taken the lead on this complex set of issues and is also working to ensure that FDA and sponsors periodically evaluate whether post-approval studies remain scientifically warranted.

To help address the various issues that have contributed to this unacceptable issue of scientists struggling to apply for federal research grants, a biomedical working group would be established to make recommendations on how scientists in this country can spend more time finding cures and less time filling out paperwork.
Title IV – Accelerating the discovery, development and delivery cycle and continuing 21st Century Innovation at NIH, FDA, CDC and CMS
Title IV includes a wide-ranging series of proposals intended to streamline regulatory processes and equip federal public health agencies with the tools they need to help accelerate the cycle of cures and foster our nation’s innovation ecosystem.
The committee has repeatedly heard about the administrative burdens scientists face in applying for federal research grants. To help address the various issues that have contributed to this unacceptable bottleneck, a biomedical working group would be established to make recommendations on how scientists in this country can spend more time finding cures and less time filling out paperwork.
Title IV also includes a series of proposals drafted by committee member Rep. Renee Ellmers that would streamline various regulatory processes companies developing new vaccines must traverse before their products can reach patients in need. Further, Title IV incorporates the hard work of the bipartisan telemedicine working group and will set the stage for new technologies to play a greater role in the delivery of quality healthcare services to Medicare beneficiaries. This team effort involved input from Pitts and full committee Ranking Member Frank Pallone, in addition to Reps. Gregg Harper, Bill Johnson, Walden, Bob Latta, Doris Matsui and Peter Welch.
Title V – Modernizing medical product regulation
Title V includes policies developed by Brett Guthrie, health subcommittee vice chairman, that would encourage drug companies to use modern manufacturing technologies here in the U.S., as well as provisions he drafted along with Shimkus intended to update certain medical device regulations, including a proposal drafted by Latta that would govern their distribution.
The 21st Century Cures committee looks forward to continuing the important conversation about how this legislation can make a meaningful difference in the lives of patients and help maintain our nation’s standing as the world leader in biomedical innovation. This discussion document is just the beginning of the legislative process on the path to cures.
Conclusion
The release of the 21st Century Cures discussion document marks a new point in the discussion, but it is one that is far from over. Prior to introduction and throughout the legislative process, the committee requests specific feedback from all interested stakeholders about how to improve the legislation. Please submit specific suggestions to cures@mail.house.gov or contact committee staff with any questions.
The 21st Century Cures committee looks forward to continuing the important conversation about how this legislation can make a meaningful difference in the lives of patients and help maintain our nation’s standing as the world leader in biomedical innovation. This discussion document is just the beginning of the legislative process on the path to cures.



