The Endolumik Gastric Calibration Tube emits near-infrared light for enhanced visibility during bariatric procedures.
Endolumik’s story started a few years ago as the FDA sounded the alarm over the risks of internal surgical staplers.
In 2019, the federal agency warned healthcare providers that it had received more than 41,000 medical device reports (MDRs) related to surgical staplers and staples for internal use from 2011 to 2018. Those MDRs tallied more than 32,000 malfunctions, at least 9,000 serious injuries and 366 deaths.
Two years later, the FDA increased the risk level associated with the staplers, reclassifying the devices from Class I to Class II and subjecting them to premarket review and special controls.
Bariatric surgeon Dr. Nova Szoka had already been working on a device that could prevent some of those injuries. She envisioned an illuminated gastric calibration tube that would shine near-infrared light through stomach tissue and fat. This camera-visible fluorescence would make it easier to see the tube inside a patient, lessening the risk of a surgeon perforating the stomach or stapling the tube to the stomach during bariatric procedures.
In 2020, Szoka founded Endolumik with Mara McFadden, now CEO of the Morgantown, West Virginia-based device developer. In 2022, they submitted their device to the FDA under the new Safer Technologies Program (STeP) and this year won the agency’s first STeP authorization.
“Safety was always the most important thing,” Szoka told Medical Design & Outsourcing.
FDA’s STeP initiative

Endolumik co-founders Dr. Nova Szoka (left) and CEO Mara McFadden [Photo courtesy of Endolumik]
STeP also offers accelerated review times, but for devices that treat or diagnose less serious diseases or conditions than the breakthrough program. The FDA’s goal for both programs is to get safer, better devices to patients faster than the agency’s traditional pathways alone.
“Advancements in medical devices that are ineligible for the Breakthrough Devices Program but offer a significant safety advantage in treating and/or diagnosing less serious diseases or conditions can also provide an important public health benefit,” the FDA said when launching STeP in 2021. “… Efforts to improve safety are directly related to improving overall clinical benefits and may also help patients experience fewer serious adverse events.”
Medical device developers can ask to join the voluntary program through a Q-Submission. Then, they’ll work with the FDA to chart the best path to marketing authorization, whether that be premarket approval, de novo or 510(k).
The FDA accepted 14 STeP requests out of 30 applications last year, according to the Center for Devices and Radiological Health’s annual report.
Taking the big STeP
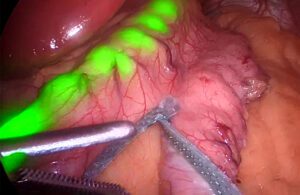
The Endolumik Gastric Calibration Tube is visible through stomach tissue and fat using a near-infrared camera. [Photo courtesy of Endolumik]
“We found them to be really responsive in terms of timely, interactive review,” McFadden said in an interview. “Sometimes you hear horror stories of submissions just sitting for the full 90 days before you hear much of anything, which was not our experience. They got back to us pretty quickly, and we had the opportunity to interactively go back and forth.”
Medical device developers should clearly understand STeP’s eligibility requirements and be prepared to discuss in depth how their device improves safety, McFadden said.
“It’s non-trivial to demonstrate that you are safer,” she said. “You have to show at least one of four different factors. You can either reduce the occurrence of a known adverse event, reduce the occurrence of a known failure mode, reduce the occurrence of a known user hazard or user error, or improve the safety of another device. You really have to do your homework and be ready to speak in depth in terms of how you’re going to have an impact on safety.”
McFadden recommended getting help from a regulatory consultant for the STeP request and 510(k) submission. Endolumik used Nilo Medical Consulting Group, led by former FDA reviewer Michael Nilo.
“The process for novel Class II medical devices is not always as straightforward as it may seem with the 510(k) program,” Nilo said. “FDA does a great job of setting up programs for innovative, safer devices that can help developers get better access to the review teams and encourage collaboration. I’d advise developers to look into all the programs and give it a shot. They are free to apply to, and the worst outcome is an introduction to the review team that you’ll be working with.”
Creating a tube with ‘Goldilocks-perfect’ balance
Endolumik’s contract manufacturer, Virtec Medical, was indispensable, McFadden said. The biggest challenge in designing the device was balancing flexibility and rigidity.
Too stiff, and the device couldn’t safely navigate through the throat and might punch a hole through the esophagus or stomach. Too soft, and the device would “buckle like a spaghetti noodle” before reaching the stomach, McFadden said.
The trial-and-error process started with the first 3D printed prototypes in pig and cadaver testing.
“Holding the balance of the functional dimensions we needed in terms of the outer diameter and the size of the electrical lumen and getting the cross-section correct to get the mechanical properties we wanted — buckling and kink resistance and that sort of thing — took more iteration than I would’ve guessed from the first go,” McFadden said. “That was one of our biggest unanticipated challenges.”
The tube is made of a PVC-like material, with one lumen for the near-infrared LEDs and another to provide suction so surgeons can remove stomach juices as needed. To determine the right material and dimensions, the team went beyond durometer testing of existing gastric tubes to find a “Goldilocks-perfect” balance.
“Our engineering team really broke down different characteristics of this device: kink pressure, buckle pressure, twisting pressure — basically quantifying all those things and then dialing it in,” she said.
At the final cadaver lab, the team invited some surgical residents to give it a try.
“Once I was able to see some trainees — who had never used a device before — safely introduce it, that’s the point where I felt we were ready to jump to a human-grade device in a clinical trial.”
Endolumik’s clinical trial has treated 22 patients so far without any adverse results, Szoka said.
What’s next for Endolumik
Endolumik’s florescent gastric tube is a single-use device. One goal for future improvements is to design for sustainability, including improved recyclability.
And while McFadden declined to go into detail about other possibilities for the company, she referenced the growing number of near-infrared cameras in operating rooms that could give surgeons better visibility with Endolumik devices of the future.
“It’s really prompted us to start thinking long and hard about where else in the world of minimally invasive surgery are there hazards created by not having a great line of sight to a tool,” McFadden said. “We definitely have one other product in the R&D pipeline that we’re really excited about, that we think is solving an equal, if not greater, pain point in the anatomy.”
“We think it’s the beginning of a digital surgical tool revolution that we’re being able to exploit the tools that are in the OR today, the towers that are in the OR today, to give surgeons advanced visuals,” she continued. “And we’re really excited to be part of what’s next as even more digital surgical tools come on the market, and to see what the next generation of cameras can do.”
Related: Proprio’s OR navigation system could improve orthopedic surgeries
![A photo of the Medtronic GI Genius ColonPro polyp detection system flagging a potential sign of colon cancer during a colonoscopy. [Photo courtesy of Medtronic]](https://www.medicaldesignandoutsourcing.com/wp-content/uploads/2024/04/Medtronic-GI-Genius-doctors-268x170.jpg)