
Many medical devices contain complex software and interdependencies with other software and hardware systems that weren’t designed to operate in a safety-critical environment. [Illustration via Adobe Stock]
A former FDA reviewer offers his perspective on building confidence in medical software validation.
By Paul Jones, Ketryx
When reviewing the software in software-as-a-medical-device (SaMD), software-in-medical-devices (SiMD) and systems-of-systems-of-medical-devices (SosMD) at the FDA, I was always looking for sufficient evidence to justify the sponsor’s claim that the device performs as intended — safely and effectively.
The arguments to support this claim are derived from the manufacturers’ quality management system (QMS) quality assurance, design controls, and corrective and preventative action (CAPA) process artifacts, which include device distribution, post-market monitoring, and updates.
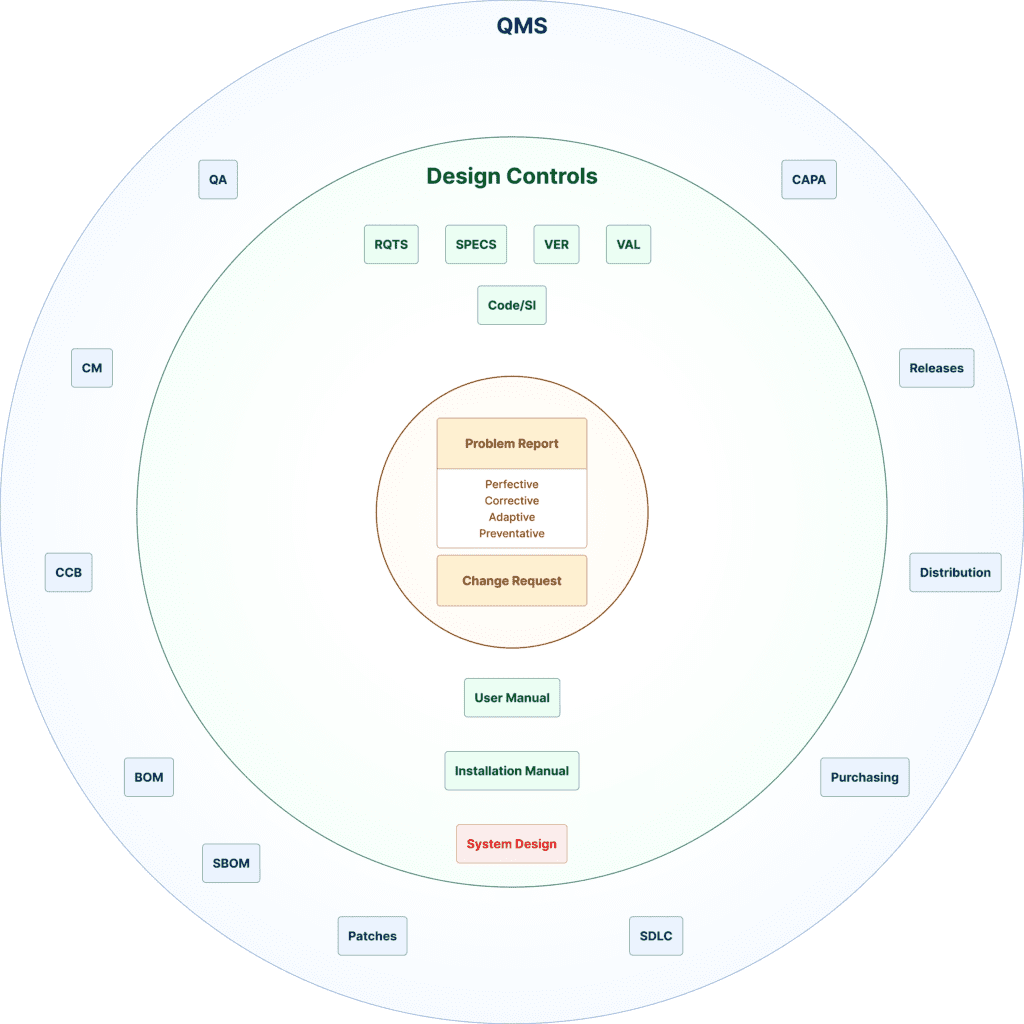
[Courtesy of Ketryx]
It starts with device requirements: what the device is supposed to do in the context of intended use, how the device will interact with its users and patients, what the environment is, and safety and security risks and mitigations. From these requirements, specifications, software development, and testing activities are performed, culminating in a validated software system.
If you can’t establish traces between the different elements of these activities, you can’t establish a credible argument that the device will perform as intended. For example, if there is no design specification corresponding to a design requirement, it is likely that this requirement won’t be included in the final product — and the device will not perform as intended.
When I was reviewing device software, I would often see poor requirement/specification properties that fell into several categories. They were ambiguous, not testable, unclear or not concise. Design control documents didn’t have unique identifiers to support traceability, were missing signatures, listed incomplete tests, lacked software quality metrics or defect/anomaly status, and had incomplete or useless traceability tables.
Too often, manufacturers didn’t use multiple complementary risk analysis methods in their risk management process. When I reviewed artifacts presented with these types of quality deficits, it was hard to have much confidence in the device behavior, and I often requested additional information (AI letter) that increased the manufacturers’ time to market.
The combination of poor quality assurance controls and poor design controls eroded my confidence in the manufacturer’s ability to adequately assure device quality over the life of the device (total product lifecycle control, or TPLC). TPLC encompasses product development, supply chains, distribution, maintenance, and end-of-life activities. If the reviewer’s confidence is low, they may reject the submission.
1. Understanding FDA guidance
On June 14, 2023, the FDA replaced the May 2005 version of Guidance for the Content of Premarket Submissions for Software Contained in Medical Devices with Content of Premarket Submission for Device Software Functions. There are major differences in the information that the FDA expects to be submitted between these two documents.
The biggest change is from a three-tiered documentation level to a two-tiered documentation level, based on risk in the context of the device’s intended use. A plethora of software-related guidance documents feed into the new software functions guidance that address multiple function device products, off-the-shelf software use, software validation, human factors, cybersecurity, and other functions and factors. Simply put, the sponsor is expected to submit the information specified in all of the relevant guidance.
The new guidance permits a declaration of conformity to the ISO 62304 standard for configuration management and maintenance practices in lieu of some specified documentation.
2. Anticipating reviewer expectations
The FDA reviewer expects to see documentation specified in all of the software-related guidances, using a checklist to verify that submitted documentation is complete. If something is missing, the review may be delayed until the documentation is provided.
In general, the software reviewer will be looking at the submitted documents for evidence of a good quality system, that known and foreseeable risks are addressed, and that the software is validated in line with the requirements of ISO 62304 and ISO 14971.
Cybersecurity-related guidance adds considerably more documentation expectations from the reviewer. FDA reviewers are increasingly devoting more scrutiny to cybersecurity concerns both pre- and post-market. Device manufacturers are relying more and more on the Internet, cloud computing, and networks to provide innovative patient services. The threat of cybersecurity events has led to an increased awareness of the importance of having a strong quality TPLC.
Vulnerabilities may be introduced anywhere in TPLC process. It is crucial to be able to maintain traceability within the supply chain, product development, production, distribution, and updates. Inconsistencies can result in harm to patients and health ecosystems.
3. Preparing your submission
Many medical devices contain complex software and interdependencies with other software and hardware systems that weren’t designed to operate in a safety-critical environment. This complexity makes it difficult, if not impossible, to rely on manual activities such as tracking progress in spreadsheets. The intricate nature of software development requires a sophisticated QMS that can handle the complexity.
Tasks such as documentation control, change management, verification, and validation activity have outgrown the limitations of traditional manual approaches. The solution lies in harnessing computing power to manage the TPLC process. By leveraging computing capabilities, information from the TPLC process can be systematically arranged, authenticated, verified, and readied for both regulatory submissions and customer delivery with a high level of confidence.

Paul Jones is VP of regulatory affairs/quality assurance at Ketryx. [Photo courtesy of Ketryx]
How to submit a contribution to MDO
The opinions expressed in this blog post are the author’s only and do not necessarily reflect those of Medical Design & Outsourcing or its employees.

![A photo of the Medtronic GI Genius ColonPro polyp detection system flagging a potential sign of colon cancer during a colonoscopy. [Photo courtesy of Medtronic]](https://www.medicaldesignandoutsourcing.com/wp-content/uploads/2024/04/Medtronic-GI-Genius-doctors-268x170.jpg)

